
What is MLD?
Metachromatic Leukodystrophy (MIM 250100) is a rare inherited lysosomal storage disorder. A functional deficiency of the enzyme Arylsulfatase A leads to sulfatide accumulation in multiple organs. Especially myelin sheaths (= white matter) in the central and peripheral nervous systems are severely affected. Patients suffer from progressive neurological deterioration and, if untreated, it leads to premature death.
Based on the age of symptom onset, four clinical MLD phenotypes are distinguished: late-infantile (<30 months), early-juvenile (2.5 – 6 years), late-juvenile (6 – 16 years), and adult (>16 years) MLD. Symptom onset at a younger age is generally associated with a faster disease progression and shorter life expectancy. More information about symptoms can be found here.
MLD is the ‘oldest’ leukodystrophy and was first described in the early last century. Nevertheless, more reseach is needed as many questions remain unanswered.
Who gets MLD?
MLD is a hereditary disease caused by genetic mutations. It can affect people of all ages, but the disease often manifests in childhood.
What causes MLD?
MLD is caused by mutations in the ARSA or, in rare cases, the PSAP gene. These genes are necessary for a properly functioning Arylsulfatase A enzyme. This enzyme is needed for the degradation of sulfatides. As a result, sulfatides accumulate in the body.
Is there a cure for MLD?
Currently, MLD cannot be cured. But, there are a few therapies are available. More information about treatments can be found here.
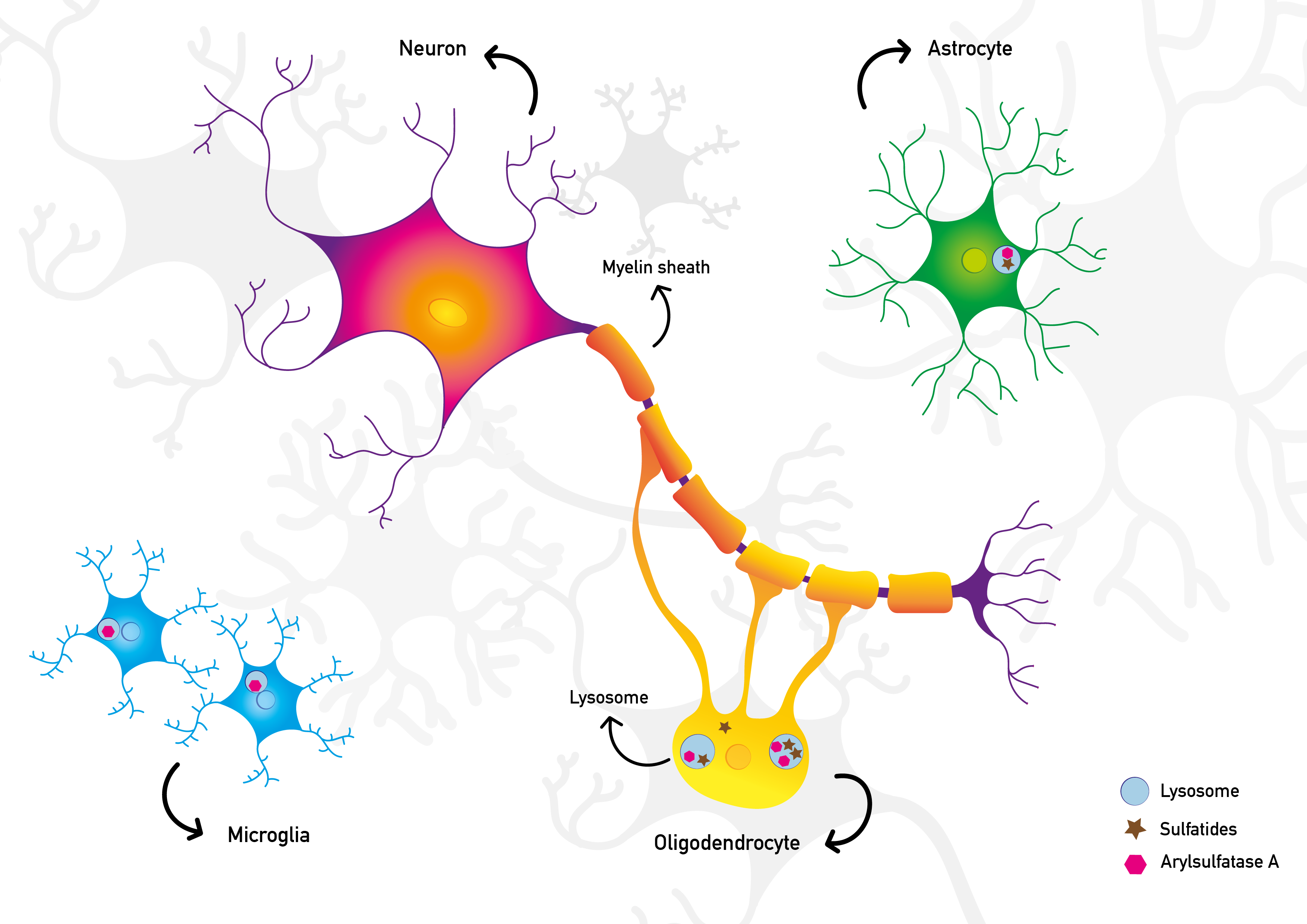

Illustration of healthy neuronal cells and the pathological situation in MLD respectively. On the first image the lysosomal enzyme arylsulfatase A is present and the sulfatides are adequately degraded. The cells in the central nervous system work normal and the myelin is intact. The second image schematically visualises the pathophysiological situation in MLD. Insufficient or absent arylsulfatase A results in the accumulation of sulfatides. Consequently, the myelin becomes severely damaged. © 2021 mldinitiative